 |
biomcmc-lib
0.1
low level library for phylogenetic analysis
|
|
biomcmc-lib
0.1
low level library for phylogenetic analysis
|
Data from alignment file. More...
#include <alignment.h>
Data Fields | |
| int | ntax |
| int | nchar |
| int | npat |
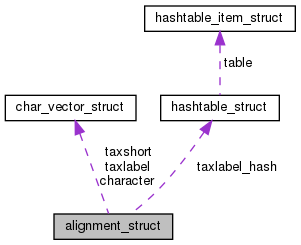
| char_vector | character |
| Number of species, sites and patterns according to sequence file. | |
| char_vector | taxlabel |
| Vector with aligned sequence for each taxon. | |
| char_vector | taxshort |
| Taxon names from file. | |
| hashtable | taxlabel_hash |
| Alias (short version) for taxon names that can be used in newick trees. | |
| int | n_charset |
| Lookup table with taxon names. | |
| int * | charset_start |
| Number of gene segments (ASSUMPTIONS BLOCK). | |
| int * | charset_end |
| bool | is_aligned |
| Start and end of each gene segment (from 1...NCHAR) (ASSUMPTIONS ). | |
| int * | site_pattern |
| FASTA files don't need to be aligned; NEXUS files do. | |
| int * | pattern_freq |
| pattern, in alignment_struct::character, to which original site belongs. | |
| char * | filename |
| if sequences are aligned, this is the frequency of each pattern. | |
| int | ref_counter |
| name of the original file, with extension removed | |
Data from alignment file.
 1.8.13
1.8.13